FlowCodeUnmix is intended to be a complete debarcoding and unmixing pipeline for spectral flow cytometry samples using the FlowCode protein epitope barcoding technology.
FlowCodeUnmix is provided under an AGPL3 licence.
To read about FlowCodes see the Immunity paper by Bricard et al. and also the ProCode system upon which FlowCodes are based.
Installation
You will need to install the following packages:
- BiocManager
- flowCore
- FlowSOM
- flowWorkspace
- remotes (or devtools)
- AutoSpectral
Everything else should be installed automatically.
Run the code below on your machine to set everything up.
# Install Bioconductor packages
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("flowWorkspace", "flowCore", "FlowSOM", "remotes"))
# You'll need devtools or remotes to install from GitHub.
remotes::install_github("DrCytometer/AutoSpectral")
remotes::install_github("DrCytometer/FlowCodeUnmix")FlowCodeUnmix is written in R and should work on any system. R, however, is not very fast, so the per-cell unmixing part will be slow. To speed this up, install the Rcpp version available at FlowCodeUnmixRcpp. This is approximately 100x faster.
Required inputs
You will need these pieces of information:
- Single-stained control samples, preferably cell-based
- A “backbone”” control sample stained with all the anti-epitope antibodies (and nothing else)
- Unstained control cells matching the source used in the backbone control
- Unstained control cells matching the source(s) in your fully stained samples
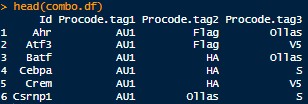
- A CSV file describing the valid combinations of epitope barcodes and what they correspond to. Example format below:
Help
FlowCodeUnmix is based around AutoSpectral. Please see the help pages and articles for AutoSpectral as a starting point. In particular, see the AutoSpectral Full Workflow.
For FlowCodeUnmix, see the example workflow in “FlowCode_Unmixing_Workflow.Rmd”, which is also available on the web at FlowCode Workflow. You can view all of the functions available in FlowCodeUnmix here.
Examples
Here is what the unmixing looks like for the backbone control (FlowCode epitopes only) with standard OLS unmixing or using FlowCodeUnmix: 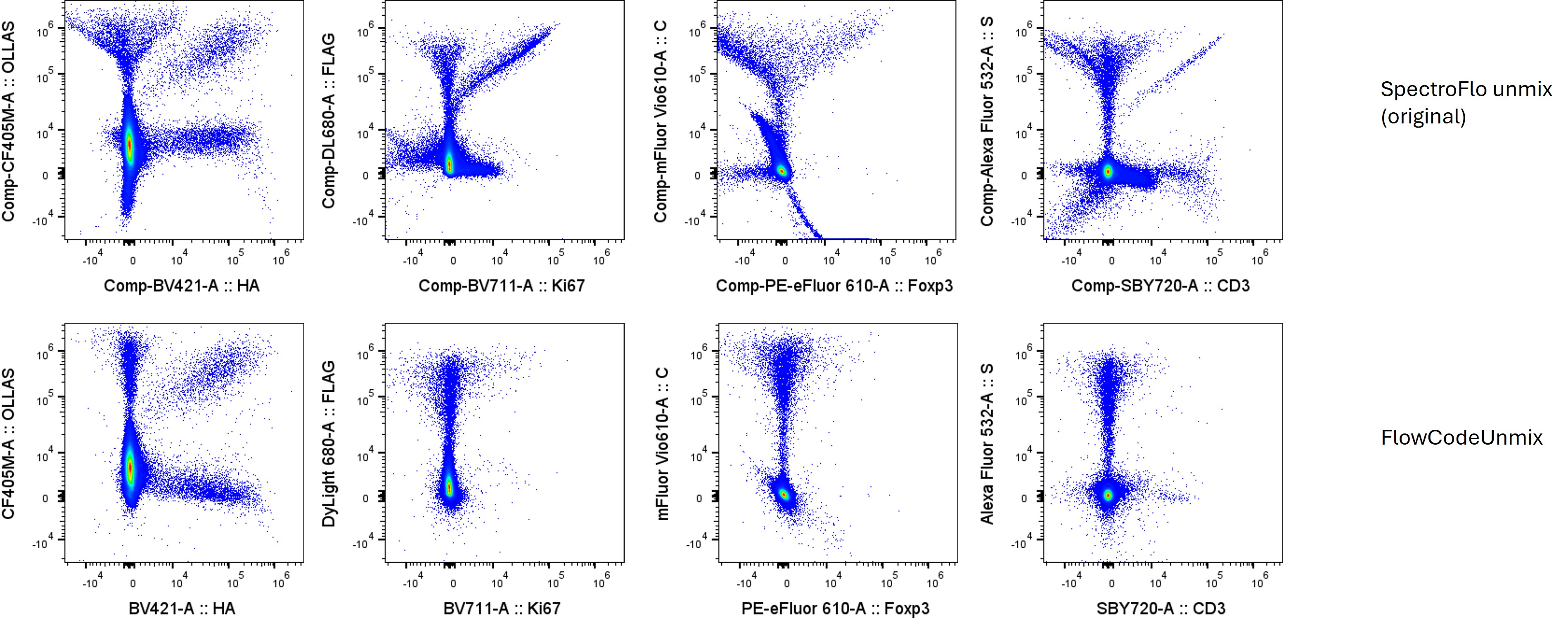
You get debarcoded channels in the output FCS file: 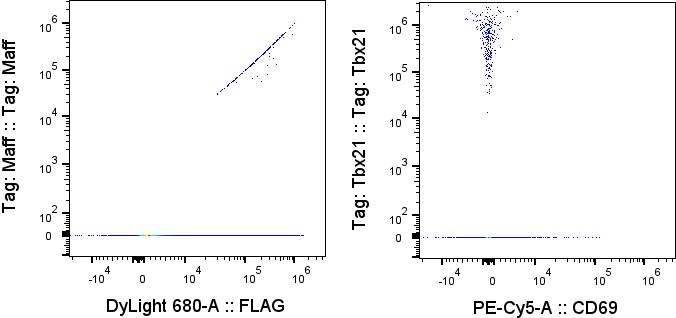
How it works
What the code does in the unmixing: * We unmix quickly with weighted least squares (WLS). * We do a first pass estimation of each cell’s best fit autofluorescence signature using minimization of fluorophore signal (worst case scenario) as the metric. * We debarcode, which tells us the best-fitting FlowCode combination (or lack thereof) per cell. * We then assess which fluorophores are present (above pos.thresholds) for a given fluorophore. If none, we return early. AF is always considered to be present (pos.thresholds for AF is -Inf). We drop down into this reduced fluorophore space for the optimization workflow because with fewer endmembers, we have more information in the residuals that we can use to identify where each cell lies on the multi-dimensional distribution. * If a cell is a FlowCode cell (has.flowcode), we need to perform a correction for FRET. This is done by aligning the residual to the deltas (fret.delta.list, fret.delta.norms) of the FRET spectra (combo.fret) for the FlowCode combination (flowcode.ids) present in that cell. This is done using a reduced set of fluorophores in the spectra for the cell, based on which fluorophores are positive (pos.thresholds), and additionally excluding any flowcode.fluors that are not present in the identified combo for the cell (using flowcode.combo.logical). We test k variants of FRET spectra (creating a new unmixed channel). The variant that best reduces the residual is selected. The projected FRET from this variant is subtracted from the cell’s raw.data (note this likely requires copying raw.data rather than just viewing it) prior to running the next steps. We re-unmix using the FRET-subtracted raw.data to re-establish estimates for fluorophore and AF coefficients. * For all cells, we next reassess the autofluorescence (AF). We identify which AF signatures (af.spectra) has been selected for a given cell based on af.idx, and construct combined.spectra using the baseline fluorophore signatures and this AF spectrum. We unmix using the reduced set of positive fluorophores as spectra, then assess which AF spectrum (af.spectra) best aligns with the residual. We test k variants for AF, select the one that gives the lowest residual, reunmix and re-establish which fluorophores are positive. We store the best AF spectrum in the final spectra. * We then assess fluorophores in the same way as for AF. We do this for each cell for positive fluorophores only, in order from greatest to least signal. We check for alignment of each fluorophore’s variants with the residual when the cell is unmixed in the reduced space (positive fluorophores only). We test k variants and pick the best, storing it in the final spectra. We track the best residual so we don’t have to re-unmix at the end of each fluorophore’s loop. * Finally, we unmix with the final spectra, which contain all fluorophores. Spectra for positive fluorophores and AF will have been optimized; spectra for negative fluorophores will remain untouched. * FlowCode and tag-specific channels are created using the debarcoded cell IDs.